{kind=link}
A major, long-anticipated regulatory overhaul has officially reshaped the compliance landscape for the European pharmaceutical and animal health sectors. EMA has removed the costly trial-and-error approach that previously slowed down veterinary peptide approvals which adversely affected the approval timelines.
Effective June 1, 2026, the European Medicines Agency’s (EMA) “Guideline on the Development and Manufacture of Synthetic Peptides” (EMA/CHMP/CVMP/QWP/367182/2025) has entered into full legal force across all European Union member states.
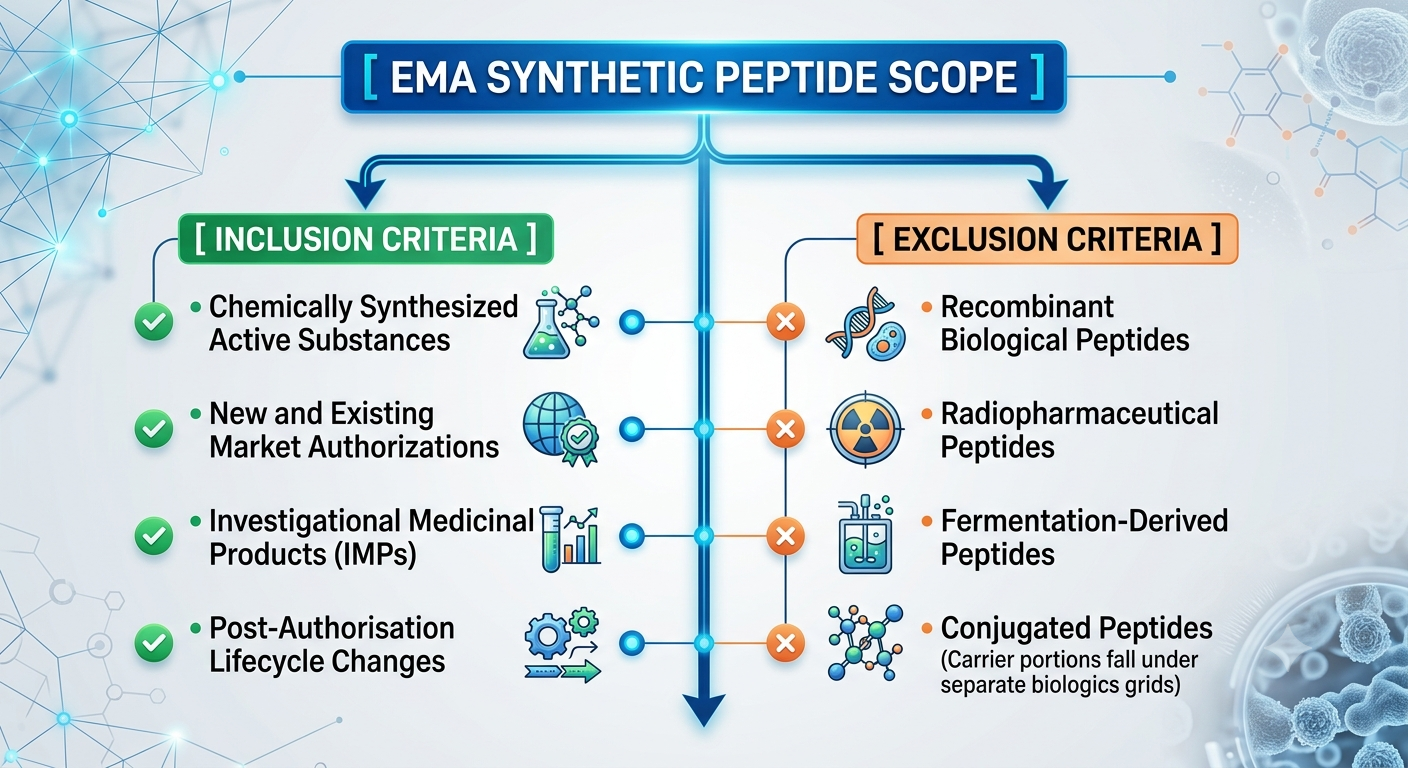
Adopted via a joint cross-disciplinary alliance between the CHMP (Committee for Medicinal Products for Human Use) and the CVMP (Committee for Veterinary Medicinal Products), the document establishes a unified, product-specific quality architecture spanning the entire lifecycle of synthetic peptide active substances.
By standardizing manufacturing controls, purification metrics, and impurity profiling, the EMA has closed a critical regulatory gap that previously forced peptide developers to navigate fragmented, non-specific chemical synthesis directives.
Closing the Regulatory Gap for Advanced Therapeutics
Peptide-based therapeutics—compounds generally consisting of amino acid chains linked by peptide bonds—have witnessed an unprecedented surge in clinical deployment. In veterinary medicine, synthetic peptides are driving next-generation innovations, including targeted immunotherapies, specialized metabolic modulators, and precise antimicrobial alternatives designed to bypass traditional antibiotic resistance channels.
Historically, because peptides sit at the structural intersection of small-molecule chemicals and complex biological proteins, regulatory submissions faced inconsistent review parameters. The new guideline replaces this ambiguity with concrete, mandatory quality expectations.
The mandate explicitly covers all new marketing authorization applications, existing substance updates, post-authorisation variations, and Investigational Medicinal Products (IMPs) utilized in clinical and veterinary field trials.